BAM file filteing to remain best isoform
Bioinformatics Asked by user977828 on January 10, 2021
I ran HiSat2, MarkDuplicate, removed reads with the lower quality score than 40 and finally only kept properly paired reads.
After the BAM filtering steps, I used the Scallop results with TransDecoder. Next, I used bamCoverage to create RNA-Seq profile. I noticed that not always the first isoform is the best one compared to the RNA-Seq profile as could be seen below a few examples:
1.

2.
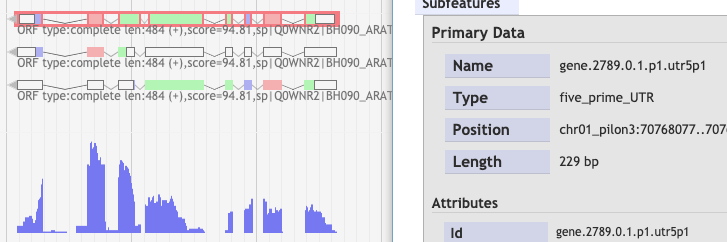
3.

4.

5.

By any chance, is there a way to keep only the best isoform which is in red box?
Thank you in advance,
Add your own answers!
Ask a Question
Get help from others!
Recent Answers
- Jon Church on Why fry rice before boiling?
- Joshua Engel on Why fry rice before boiling?
- Peter Machado on Why fry rice before boiling?
- haakon.io on Why fry rice before boiling?
- Lex on Does Google Analytics track 404 page responses as valid page views?
Recent Questions
- How can I transform graph image into a tikzpicture LaTeX code?
- How Do I Get The Ifruit App Off Of Gta 5 / Grand Theft Auto 5
- Iv’e designed a space elevator using a series of lasers. do you know anybody i could submit the designs too that could manufacture the concept and put it to use
- Need help finding a book. Female OP protagonist, magic
- Why is the WWF pending games (“Your turn”) area replaced w/ a column of “Bonus & Reward”gift boxes?